Click the buttons below to try the website yourself!
nETPHOS 2.0

NetPhos 2.0 is a website that determines the phosphorylation sites on any given sequence or sequences, and rates them on a scale of 0-1 with anything over the .5 line being significant and an active site of binding. This website can handle multiple sequences at once and is used with protein FASTA format such this:
- >Homo Sapiens 1433 sigma MERASLIQKAKLAEQAERYEDMAAFMKGAVEKGEELSCEERNLLSVAYKNVVGGQRAAWRVLSSIEQKSN EEGSEEKGPEVREYREKVETELQGVCDTVLGLLDSHLIKEAGDAESRVFYLKMKGDYYRYLAEVATGDDK KRIIDSARSAYQEAMDISKKEMPPTNPIRLGLALNFSVFHYEIANSPEEAISLAKTTFDEAMADLHTLSE DSYKDSTLIMQLLRDNLTLWTADNAGEEGGEAPQEPQS
mEME

MEME is a website that identifies conserved regions within a protein or between multiple protiens called motifs. Motifs are short, conserved regions of the protein that allow the domain to carry out a necessary function. They also described the potential secondary structure of the protein. They are also generally the most highly conserved parts of the genome. It has an output of various HTML visual files, such as the picture about and it gives an idea of where the most conserved regions of any protein are. This program also uses protein FASTA format, as shown above in the NetPhos 2.0 description.
blast

BLAST or Basic Local Alignment Search Tool is used to align proteins with its homologs among other organisms. It outputs an image similar to what is above and also provides a list of different proteins. These proteins could all be from the same organism, a different organism, or many different organisms. This flexibility allows the researcher to see how identical the homologs are between organisms, and how conserved the proteins are among organisms. An example of how to use this information is found here. This aids in determining the exact homology of your protein. It can also be run with gene FASTA or protein FASTA.
Go: gene ontology

Gene Ontology or GO is broken down into 3 groups that describe the categories that define a gene and what is does in the cell. These terms are biological processes, cellular components and molecular function. It is comprised of numerous databases and provides insight into what role the gene or protein has within the cell. This website is very easy to use and the name of a gene or protein is all you need to begin observing the function, localization and biological necessity of your protein.
This Ontology was done using Gene Ontology Consortium, but you can also access this information on UNIPROT or ENSEMBL.
This Ontology was done using Gene Ontology Consortium, but you can also access this information on UNIPROT or ENSEMBL.
smart
|
PFAM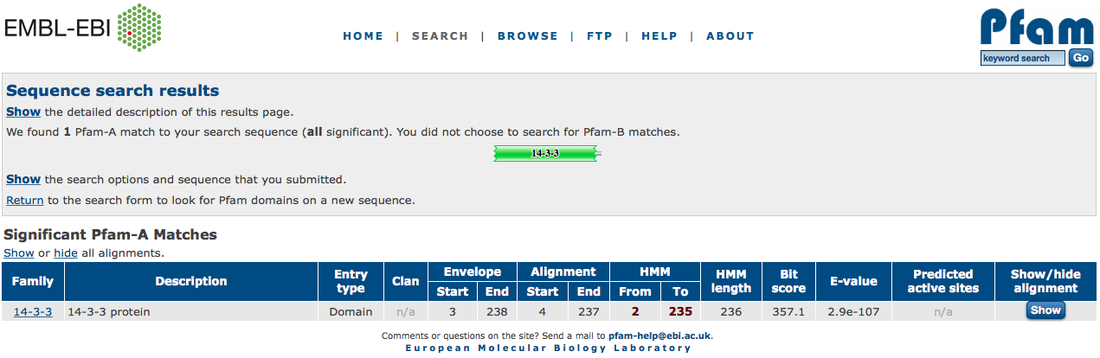
|
SMART and PFAM are protein domain exploration sites. They both have generally the same function, however I liked SMART better because it had more usable features, they gave you more information as to what the domain did, how long it was, what pathways it is involved in and where the post-translational modification sites are on the domain. PFAM is not quite as fully functional, but it is necessary to cross reference these two against each other because there can be discrepancies in the literature that don't account for changes in domains or the number of domains.
crispr-cas9
CRISPR or Clustered Regularly Interspaced Short Palindromic Repeats are portions of prokaryotic DNA that can be used to create transgenic line and basically "copy and paste" any sequence that you want into an already formed gene. This is performed with the help of the cas9 gene and guide RNAs to chop and insert the desired DNA sequences into the genome. The video above explains very well how this is the future of scientific discovery.
uniprot-SUPER USEFUL!!

UniProtKB is probably the most useful site in this entire project because it describes everything you need to know about your gene and protein, and includes gene ontology, taxonomy, post-translational modifications and pathways. If the information is lacking in any particular area that you need covered, it will generally link out to a more specialized data base.
Clustal Omega

Clustal Omega is another multi-functioning bioinformatic websites, it provides more sequence comparisons among conserved regions. This website however also provides more information on phylogeny. After you insert your various sequences, (generally different organisms) you can create a phylogenetic tree with a variety of different matrices. These are describe in greater detail here.
String
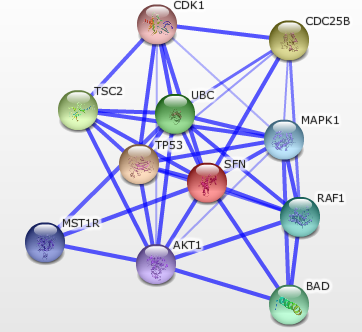
STRING is an information database that function in determining known and predicted protein-protein interactions between 1 or many proteins. It can be used in a variety of ways and the more advanced functions will help determine which protein is affecting which or if they are working together. You can also click on each of the colored circles to see what the function of each protein is and how it might interact with your desired protein. It also links out to UniProtKB and your model organism database so you can view more information on your protein interactions. You can see a larger more in depth visual of STRING here.
